FDA将在60天内决定是否接受申请并展开审评。
美东时间2020年7月8日上午,渤健(Biogen)宣布与其合作伙伴卫材(Eisai Co.,Ltd.)针对其治疗阿尔茨海默病(AD)的治疗药物Aducanumab,已完成向美国食品药品监督管理局(FDA)提交生物制品许可申请(BLA)。
新闻稿中称,“一旦获批,Aducanumab将成为首个能够减缓阿尔茨海默病患者临床衰退的疗法,并将成为首个证明清除β-淀粉样蛋白可以带来更佳临床结果的疗法。”
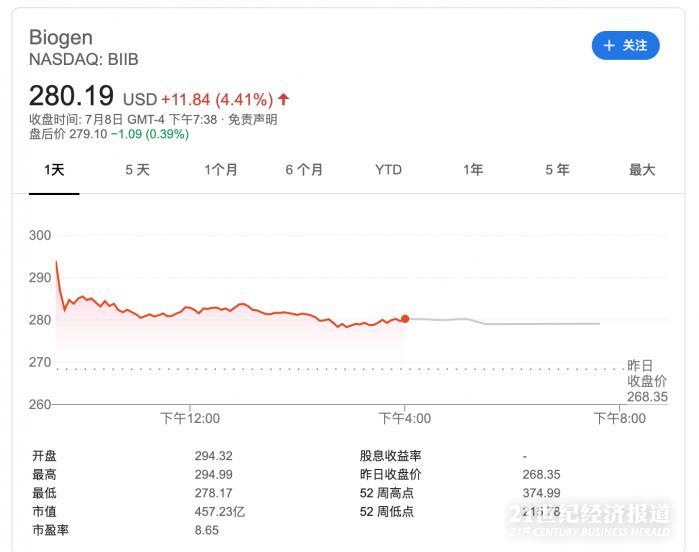
受此消息提振,周三Biogen股价上涨4.41%,每股收至280.19美元。
这个似乎是阿尔茨海默症药物研发的“重型炸弹”来自去年震惊业界的那项“起死回生”的临床试验,因为此前这已经是一项被判“死刑”的临床试验。
2019年3月21日,Biogen宣布终止Aducanumab两项代号分别为ENGAGE和EMERGE的全球III期研究,独立委员会评估其很可能难以达到预期疗效。其时,Biogen股价应声大跌27%,市值缩水150亿美元。而10月底再申请消息一出,Biogen股价迅速大涨40%,3月份丢掉的150亿妥妥收了回来,市值攀至520亿美元。
FDA会批准这个临床跌宕起伏、效果迷雾重重的药物吗?
复盘“起死回生”
Biogen要申请上市的药物Aducanumab(BIIB037)是一款研究用人单克隆抗体,用于早期阿尔茨海默病的治疗。根据此前的合作开发和许可协议,Biogen获得Neurimmune授权许可引入了Aducanumab。自2017年10月以来,Biogen和卫材在全球范围内合作开展了该单抗药物的开发和商业化。
此次申请提交包括了III期EMERGE试验、ENGAGE试验,以及1b 期PRIME试验的临床数据。在此项申请中,Biogen同时申请优先审评资格。一旦获批,Aducanumab将成为首个能够减缓阿尔茨海默病患者临床衰退的疗法,并将成为首个证明清除β-淀粉样蛋白可以带来更佳临床结果的疗法。
Aducanumab临床开发计划包括两项针对早期阿尔茨海默病患者的III期临床试验:EMERGE和ENGAGE,入组因阿尔茨海默病而患有轻度认知功能障碍(MCI)的患者以及简易智能状态量表(MMSE)评分为24-30的轻度阿尔茨海默病痴呆症患者。
EMERGE和ENGAGE为两项III期多中心、随机、双盲、安慰剂对照、平行组试验,旨在评估Aducanumab的疗效和安全性。这两项研究的主要目的是评估每月使用Aducanumab与安慰剂相比在减少患者认知与功能障碍方面的疗效,衡量指标为CDR-SB评分的变化。次要目标是评估每月使用Aducanumab与安慰剂相比在降低患者临床衰退方面的疗效,衡量指标为MMSE、ADAS-Cog 13和ADCS-ADL-MCI。
2019年3月21日,根据此前基于早期、小规模数据集的预先设定无效性分析结果,这两项研究终止,宣告失败。
在3月份决定终止试验时,主要分析的数据是截至2018年12月26日的可用数据,共涉及1748例有机会完成18个月研究期的患者,无效性分析的结果显示,两项试验可能无法在完成时达到其主要终点。
但试验终止之后,研究人员获得了更多新数据,包括3285名患者(其中2066名患者有机会完成18个月的治疗期)。基于这个规模更大的数据集,Biogen认为,“EMERGE在预先设定的主要终点上具有统计学意义(P = 0.01)。尽管ENGAGE试验并未达到其主要终点,但其中一类亚组患者的相关数据为EMERGE试验的结果提供了支持。”
在EMERGE试验中,基于预先设定的次要终点指标,高剂量Aducanumab组患者的临床衰退同样持续减缓。淀粉样斑块沉积成像结果显示,与安慰剂相比,低剂量与高剂量Aducanumab在26周和78周时均能减轻淀粉样斑块负担(P <0.001)。尽管ENGAGE(1647例患者)没有达到其主要终点,但Biogen认为,来自ENGAGE的一个子集数据对EMERGE试验的结果提供了支持。
在这两项研究中,最常报告的不良反应事件是淀粉样蛋白相关的影像学异常水肿(ARIA-E)和头痛。大多数ARIA-E患者在ARIA-E发作期间没有出现症状,并且ARIA-E发作通常在4至16周内消失,通常没有长期的临床后遗症。
Biogen就这些不同的结果及其影响向外部顾问和FDA进行了咨询。
Aducanumab的临床计划还包括1b期PRIME研究及其对早期阿尔茨海默病患者的长期扩展研究(LTE)(入组前驱期阿尔茨海默病患者或MMSE评分为20-30的轻度阿尔茨海默病痴呆症患者)。这项研究的结果表明,Aducanumab对于β-淀粉样斑块负担的减轻呈现时间和剂量依赖性,同时针对探索性临床终点的分析显示出了临床衰退的减缓(10毫克/公斤,在治疗12个月时CDR-SB和MMSE均具有统计学意义的改善),并且在为期48个月的长期扩展研究中持续获得减缓。
据Biogen方面称,“临床数据显示,Aducanumab有望影响疾病的病理生理机制,减缓认知和功能的衰退,改善患者的日常生活能力,包括进行个人理财、家务活动(如打扫卫生、购物和洗衣服)和独自出门旅行。”
在生物制剂上市许可申请提交前,Biogen称与FDA“如期进行了上市申请前会议。FDA将在60天内决定是否接受申请并展开审评。如果接受申请,Biogen预期FDA同时能告知该申请是否被授予优先审评资格。 随后,Aducanumab的生物制剂上市许可申请将接受FDA的审评以决定能否获批。”
除向FDA提交生物制剂上市许可申请外,Biogen表示还继续与包括欧洲和日本的其他监管机构进行对话,努力实现在这些市场中提交申请的目标。
FDA会批准吗?
渤健全球首席执行官冯纳玺在新闻稿中说,“阿尔茨海默病是当今最大的公共卫生挑战之一。它让我们所爱的人逐步失去记忆力和独立生活的能力,最终剥夺他们的基本行为能力。Aducanumab是首个向FDA提交的针对此严重疾病的相关临床症状衰退和病理机制的生物制剂上市许可申请。我们致力于推动阿尔茨海默病领域的医学进步,期待FDA对我们的申请进行审评。”
但,FDA会批准吗?
阿尔茨海默病即Alzheimer's disease,简称AD,俗称“老年痴呆”。痴呆症患者有多种类型,阿尔茨海默病占比最大,约占所有病例的60-70%。
阿尔茨海默病是一种进行性神经系统疾病,会损害患者的思维、记忆力和独立性,导致过早死亡。比如频频见诸于报端的老年人迷路走失,这类记忆减退、障碍是AD患者最典型的症状之一。
该疾病目前无法阻止、延迟或预防,影响着疾病患者及其家人。根据世界卫生组织(WHO)的数据,全世界有数千万人患有阿尔茨海默病,这一数字在未来几年还会增长,从而使得疾病所需的医疗资源出现不足,花费将高达数十亿美元。
阿尔茨海默病的特征在于大脑的变化,包括毒性β-淀粉样斑块的异常积聚,这种积聚始于患者出现疾病症状前约20年。由阿尔茨海默病引起的轻度认知障碍是疾病的最早阶段之一。在这一阶段,症状开始变得更加明显,并可以被发现和诊断。目前的研究工作专注于患者的早期发现和治疗,以最大程度地减缓或阻止阿尔茨海默病的进展。
阿尔茨海默病除了是一种至今无法治愈的疾病,也是全球药物研发史上成功率最低的领域。
截至目前,美国FDA批准治疗阿尔茨海默病的药物共有五个,包括多奈哌齐、加兰他敏、美金刚和卡巴拉汀等。但自2003年美金刚获批上市,过去十五年,真正意义上的新药至今没有再出现,研发失败率高达几乎百分百。
阿尔茨海默病除了是大小药企们临床试验的大型“坟场”,对于患者来说,更是面临着“无药可治”的境地。由于阿尔茨海默病的发病原因和机制至今尚未明确,现有的药物和认知行为治疗仅有助于减缓症状,而没有治愈的方法,也没有有效的可以减缓疾病进程的药物。
“老年痴呆”的研发天空一片阴霾,是跨国药企们折戟的大型战场。对于此次渤健的“起死回生”,监管机构会怎么做?
“这里面肯定还是有一些不确定性的,类似失败的临床试验再申请这样的操作,在我印象中是没有的。”北坡生物科技有限公司创始人李文凯此前在接受21世纪经济报道采访时表示,对于新的分析结果,“ENGAGE的数据可能不太有说服力,EMERG会更重要一些,实验更加完整。”
由于常年没有新药上市,也会影响监管机构对于审评审批的态度。
“一是最近几年FDA放松了一些药品要求,尤其是像AD这样很长时间没有新药的疾病,面临一些压力;二是制药行业一直在游说,因为大家烧了这么多钱,到目前为止还没有真正的有效药物;三是AD这个病本身给美国社会造成了很大的负担;另外在美国患者组织是很有影响力的,美国社会对于阿尔茨海默病的认知度非常高,美国有国家性阿尔茨海默症法案,在奥巴马时期通过,所以不管从政策、舆论还是民意方面,都有比较大的推力,就是希望能够让有可能改变疾病进程的或者有一定治疗效果的药物能够早点上市。”李文凯分析,“所以在这种背景下,我觉得是存在这样的可能性:FDA审批的时候,尤其是专家委员会大部分投票支持,获批的可能性比较大。”
淀粉样蛋白假说的生与死
研发阿尔茨海默症的药企巨头们终于能从“坑里”爬出来了吗?阿尔茨海默症终于要迎来十七年“寒冬”后的春天了吗?
另一家在阿尔茨海默“坑里”耕耘了30年的制药公司是礼来,不过“屡战屡败”的礼来今年以来在阿尔茨海默研发上可以说喜忧参半。
2月10日,礼来宣布对通过华盛顿大学的显性遗传阿尔茨海默DIAN-TU研究表明,其阿尔茨海默症药物solanezumab不符合主要终点。DIAN-TU平台试验是第2/3期随机、双盲、安慰剂对照研究。目的是测试由稀有基因突变引起的罹患阿尔茨海默氏病或处于遗传性阿尔茨海默氏病的风险中的潜在疾病改良疗法。
Solanezumab也是一种研究型抗淀粉样蛋白单克隆抗体,正在无症状阿尔茨海默氏症的抗淀粉样蛋白治疗中用于临床前研究。这项临床试验,测试的是solanezumab用于年龄较大、有淀粉样蛋白,但未显示记忆障碍的症状的患者。
好消息是,5月28日,FDA批准了礼来的TAUVID™用于阿尔茨海默病患者的诊断,适用于大脑的正电子发射断层扫描(PET)成像,以评估阿尔茨海默病的成年认知障碍患者中聚集的tau神经原纤维缠结(NFT)的密度和分布。
“对抗AD的斗争需要对该病的两种关键病理进行精确和可靠的评估,因为仅仅通过临床评估来准确诊断病人是有限的。”礼来公司疼痛和神经退行性病变研究开发副总裁Mark Mintun医学博士说,“随着FDA批准AMYVID来证明这两种病理之一——淀粉样斑块的存在,创造了历史。我很高兴TAUVID现在已经被批准用于成像tau NFTs,这是另一个关键病理,可以对患者进行更全面的评估。”
如果渤健的Aducanumab成功“起死回生”,还有对于研究层面更深层次的影响。“他们要么会复兴淀粉样蛋白β理论,要么彻底杀死它。”
阿尔茨海默病的发病原因不明,但在过去三十年人们对它的认知有很大的发展。
礼来中国医学总监吴胜虎此前在接受21世纪经济报道专访时指出,“从1987年开始,学界建立了建立了基于β淀粉样蛋白假说和Tau蛋白假说的理论基础,开发了相关的靶点进行药物研发。”
目前全球各大公司的研发管线几乎都围绕这两个理论基础进行通路开发。虽然这些疗法虽然能够成功降低Aβ水平或者消除大脑中β淀粉样蛋白的沉积,但是对患者的认知能力的衰退却没有帮助。
由于失败案例实在太多,从学术到临床到药物都对β淀粉样蛋白假说不断提出质疑。如 BACE和Aβ这两个靶点都是各家失败的重灾区,目前也没有成功的产品出现。
由于理论基础长期没有更大的突破和新的学说,也是临床成功率太低的原因之一。
β淀粉样蛋白假说是够无效?在基础研究领域还有哪些进展?
“近几年的进展还包括神经影像学的应用、越多生物标志物的使用,比如认识到脑部的老年斑蓄积、缠结。”礼来中国临床研究医师成燕此前在接受21世纪经济报道采访时表示,现在对AD的诊断也会越来越早期和细分,“现在需要更早地去探测没有症状或症状很轻微的病人,因为业界都意识到目前的临床试验都干预得太晚了。”
2018年2月,FDA新发的一份针对AD新药研发的指南中,也建议将AD治疗药物所针对的疾病进程调整为早期阶段。
诊断的复杂性也是临床试验失败率高的一大因素。“老年患者入组临床试验本身就是比较困难的,除了阿尔茨海默病,他可能还有其他各种身体问题,或者不同的病理但是临床表现可能相似,如何分组、比较也有一定的难度,加上慢性病的临床试验期很长,投入巨大,耗时费力。”李文凯认为,虽然针对淀粉样蛋白假说来做临床开发的公司很多,但每家具体做药物开发是不太一样的,包括临床实验设计、入组标准,测量指标等,“大部分会比较相似,但差异会导致每一家做出来结果不同。渤健的的临床试验设计是比较早的,尤其是把一些重要的生物标志物纳入了入组标准,并且入组的病人相对症状较轻。因为淀粉样蛋白的变化是发生得非常早的,等到病人出现症状的时候,可能病理已经在他大脑里发展了二三十年了。”
“我认为渤健这个药物即使获批,效果也不会特别显著。”李文凯认为,但如果获批,会对其他做同类研发的公司有很大的鼓舞,理论上讲其他家也完全有可能成功。
资本游戏还是患者福音?
阿尔茨海默病的临床试验失败简直已经是各大药企的家常便饭,但制药巨头们的热情风吹不灭、雨打不熄,几十个亿的美金源源不断往里扔。
2019年1月30日,罗氏宣布终止crenezumab 治疗早期阿尔茨海默病患者(前驱或轻度AD患者)的两项III期CREAD I和CREAD 2临床研究。罗氏做出此决定同样是因为独立数据监测委员会对于其疗效的担忧。
2018年6月,阿尔茨海默研发领域的老大哥礼来和阿斯利康宣布终止了治疗阿尔茨海默病的全球III期临床试验项目lanabecestat(BACE抑制剂),这是继默沙东宣布终止其III期项目verubecestat、强生宣布终止BACE抑制剂atabecestat 的II/III期项目后,又一个倒在阿尔茨海默病研发战场上的巨头。
同时,lanabecestat也是礼来在2016年底solanezumab(Aβ抑制剂)III期失败之后,又一次折戟在III期临床的阿尔茨海默病项目。
不停打水漂,制药公司们是傻吗?
不是。这是一个只要成功,就可以横扫市场的“重磅炸弹”,年销售额至少十亿美金起步。长期以来,阿尔茨海默症药物一直被认为是生物制药研发中最大的蛋糕,具有数十亿美元的潜力。如果Biogen现在可以说服监管机构获批,那么他们将完成行业历史上最戏剧性的转变。
由于市场的巨大空白和社会老龄化的压力,即使研发困难重重,制药公司们依旧以数十亿美元的巨额投入“前赴后继”。根据clinicaltrials.gov的注册数据,罗氏、渤健、杨森、礼来、武田、灵北等巨头的身影依旧还在。
作为阿尔茨海默研发“带头大哥”的礼来,自1988年之后,“到目前投入已经接近40亿美金,但最终真正能够上市突破性的产品,我们估计还要花至少10-20亿美金。”吴胜虎表示,由于临床失败率太高,“且III期投入大概要占到全部投入的50%,我们在去年也做出了一个转变的调整,之后希望在有更强硬的临床试验基础上,再跨入三期研究,同时也会有更多的合作和联盟。”
2018年1月,武田宣布与Denali达成合作联合开发包括两个阿尔茨海默病项目在内的神经退行性疾病项目,金额接近12亿美元。
这无疑是巨额市场的诱惑和利益驱动,据世卫组织估计,目前全球社会痴呆症的总成本每年超过6060亿美元,超过全球GDP的1%。
根据灵北公司(Lundbeck)2017年财报显示,2016年全球阿尔茨海默病的市场大约为44亿美元;而在上市近16年后,灵北认为Ebixa®(美金刚)还有7亿丹麦克朗的市场,2017年美金刚在除欧洲和北美以外的全球市场卖了0.71亿美元。在最新的2019年财报中,灵北并未单列美金刚的营收,但与其他几个药一起在全球卖了约2.33亿美元,同比增长9%。
2016年,在礼来宣布Solanezumab III期临床失败之前,华尔街的分析师们对这个寄予厚望的药物给出了一年至少十亿甚至上百亿美金销售额的预期,“如果这样的产品成功上市,毫无疑问是重磅型药物,即一年销售额至少十亿美金。由于大家的通路相同,无论哪家成功都是好事,意味着后续成功的可能性也很大。”
对于去年至今渤健的操作,资本市场飞快给出了正向反馈,但由于过于戏剧性,也让业界保持质疑。从去年三月以来,渤健面临着非常困难的局面,每况愈下,市场上坏消息多于好消息,某种程度上,非常需要正向消息的提振。
如果FDA批准,Biogen将会是未来很长一段时间内阿尔茨海默症的“王者”;但不管批准与否,寻找AD治疗的任何积极进展,即便只有1%,对患者都是积极的。
(作者:卢杉 编辑:张伟贤)