医健IPO解码|获批首个国产新冠口服药才10天,真实生物为何此时选择赴港上市?

作为一家尚未实现营收的Biotech企业,真实生物能否持续通过融资来支撑研发?刚获批上市的阿兹夫定又如何快速商业化实现盈利?
21世纪经济报道记者 季媛媛 上海报道 在核心产品阿兹夫定片附条件获批上市仅过去10天,真实生物科技有限公司(简称“真实生物”)就向港交所提交了上市申请。
8月4日晚间,港交所官网文件显示,真实生物向港交所第一次呈交了上市申请书,保荐人为中金公司。上市申请书显示,真实生物的高级管理层有7人,其中王朝阳是真实生物的创始人。真实生物在上市前的股东架构中,王朝阳通过三联创投、Creative Summit(受限制股份单位计划),分别持有46.89%、1.72%的股份,合计持股48.61%的股份;杜锦发博士,通过Modern Target持股17.55%。
不过,上市申请书并未披露王朝阳的具体履历,仅显示,王朝阳是公司控股股东之一,2019年9月26日获委任为贵公司非执行董事,并均于2022年8月1日辞任。另据公开报道,王朝阳是平顶山人,早期曾在多个领域创业并收获了他人生的第一桶金。2011年,王朝阳从发明人常俊标手中购买当时还是一种化合物的阿兹夫定的专利。
据公开资料,河南真实生物科技有限公司成立于2012年9月12日,公司经营范围主要包括研发、生产和销售抗癌药、抗病毒药、糖尿病相关疾病治疗药和心脑血管及衰老相关疾病的治疗药。而在7月25日,国家药监局应急附条件批准真实生物阿兹夫定片增加新冠肺炎治疗适应症注册申请,用于治疗普通型新型冠状病毒肺炎(COVID-19)成年患者,该药也是首个获批上市的国产治疗新冠肺炎口服药物。
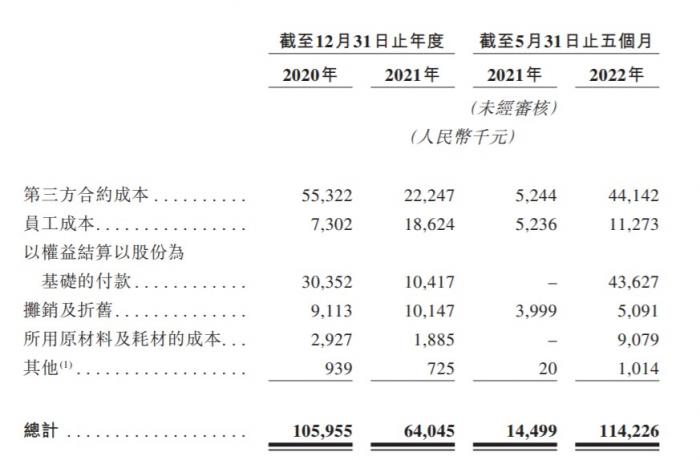
这也使得真实生物此次冲刺港股备受关注。据真实生物上市申请书披露,此次赴港上市,公司也计划将募集资金用于核心产品阿兹夫定治疗COVID-19的制造及商业化。申请书显示,在过去的2020年、2021年和2022年前五个月,真实生物未有营业收入,相应的研发开支分别为1.06亿、0.64亿和1.14亿元人民币,相应的净亏损分别为1.51亿、1.97亿和2.18亿元人民币,合计近两年多的时间内亏损达5.66亿元。
尽管真实生物对外解释称,绝大部分经营亏损来源于研发活动产生的成本、与经营相关的行政开支、财务成本及可转换可赎回优先股的公允价值变动。但不难发现,对于一家尚处于亏损的生物医药公司而言,走向资本市场也不失为一个方向。不过,作为一家尚未实现营收的Biotech企业,真实生物能否持续通过融资来支撑研发?已经获批上市的阿兹夫定又如何快速放量实现盈利?这些问题备受市场关注。
来源:上市申请书
赴港上市时机选对了?
中信证券研报显示,2022年一季度全球泛医疗健康领域投融资总额都呈现了下降趋势,其中中国投融资总金额299.97亿元,环比下降37.46%,同比下降34%左右,超过了全球降幅。在生物医药领域,据浙商证券统计,2022年4月,累计发生39例融资事件,涉及金额5.68亿元,环比下降47%。
从致同咨询观察到的数据来看,生物医药LS行业融资总额相较前两年有所下降,这是一个明显的趋势。究其根源,一方面是由于近两年宏观环境的变化带动了资本市场波动的加剧,且板块轮动、生物医药企业(LS)不再处于潮头也是影响融资总额下降的直接因素;另一方面,需要更加关注资本市场对生物医药企业估值大幅回调的现象,该等估值下降至今尚未得到足够修复,也反向导致了生物医药企业融资总额的下降。
致同咨询生命科学与健康行业领导合伙人、融资与并购财务顾问服务合伙人董慧慧在接受21世纪经济报道记者采访时指出,总体来看,我们认为,生命科学行业的参与方,不管是资方还是企业,双方仍对行业未来报有足够的信心,当前仅处于等待估值和市场修复的阶段。需要特别指出的一点是,未来资本市场在生物医药行业的投资会恢复,但关注的细分领域将发生显著的改变。
真实生物聚焦的细分赛道是病毒性疾病领域。根据弗若斯特沙利文分析,病毒性疾病是全球医药市场最大的治疗领域之一。由于城市化迅速进展及前所未有的全球互联互通,大量人口受到病毒感染的影响,加之致力于开发抗病毒药物的公司数量有限,导致大量医疗需求未得到满足。包括1980年代的全球AIDS流行及持续的COVID-19疫情在内的传染性病毒性疾病的爆发对公共卫生构成了严重威胁,并给社会带来巨大的经济负担,凸显对更有效的预防及治疗方案的需求。
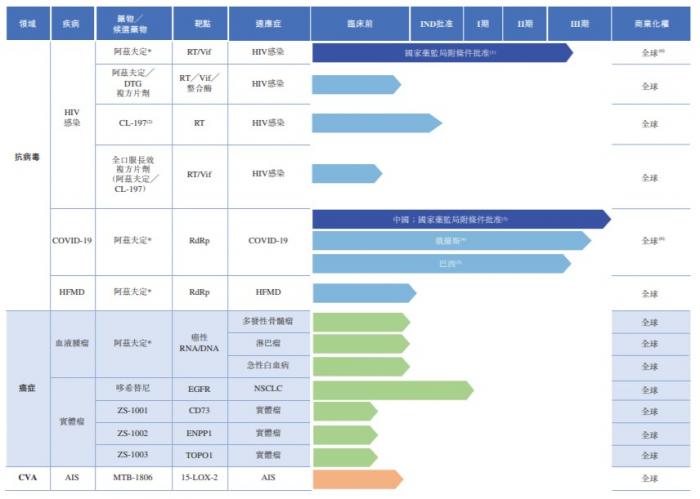
也是基于此,真实生物在抗病毒药物领域进行了布局,药物管线包括治疗HIV感染及COVID-19等的新型疗法,而核心产品阿兹夫定更是被认为具有广谱抗病毒活性的创新药物,已分别于2021年7月及2022年7月获国家药监局附条件批准用于治疗HIV感染及COVID-19。
在阿兹夫定的商业销售上面,真实生物与复星医就在中国内地及潜在的全球若干区域签订联合开发及复星医药产业独家商业化战略合作协议。招股书披露,真实生物也已经与中国多家领先药品制造商(包括北京协和)订立战略协议,为其生产及供应阿兹夫定的活性药物成分(API)及片剂。真实生物方面也强调,公司亦具备自行生产能力,年产能约为十亿片剂阿兹夫定。
来源:上市申请书
除了阿兹夫定,真实生物也在开发其他用于治疗病毒性、肿瘤及脑血管疾病的多种创新候选药物, 包括CL-197,一种用于治疗HIV的口服长效嘌呤核苷抗病毒药物;哆希替尼,一种第三代表皮生长因子受体酪氨酸激酶抑制剂(EGFR-TKI)抗肿瘤候选药物,与广泛使用的第三代EGFR-TKI相比,其有潜力成为更安全、毒副作用更低的药物;以及MTB1806,一种用于急性缺血性脑卒中的小分子候选药物,通过在临床前研究中观察,其较低剂量水平的给药方案可达到与NBP(一种国家药监局批准的AIS药物)相当的效果。此外,截至最后实际可行日期,真实生物于临床前阶段有若干其他小分子候选药物,用于治疗实体瘤。
有证券机构医药行业首席分析师在接受21世纪经济报道记者采访时介绍,真实生物瞄准的这些热门的研发领域,背后也是符合生物医药行业普遍存在的高投入、高产出、高风险、高技术密集型等特点。而从宏观层面来看,生物医药板块一直在创新高,我国的创新药IND、NDA等数量持续地增长,生物医药板块仍然是处于一个蓬勃发展的阶段,要想在热门赛道持续进行发力,各个细分板块内在增长的逻辑一直是没有变。而真实生物也必将面临诸多的竞争对手抢食的局面,如此也意味着真实生物需要更多的资金来推动后续的产品线稳步进行,这也是真实生物选择在此时间窗口赴港上市的重要原因之一。
“创新药商业化进程拉长、一些Biotech企业可能出现经营难题等挑战,我们认为这在中国生物医药产业链逐渐发展完善的过程中必然会出现的情况。与其他产业一样,只有优质且健康的企业才能在产业链中找到自己适当的位置并生存下去,但产品单一、财务和经营现金流不好的企业可能会面临破产的风险。所以,加速研发投入成为Biotech企业需要进一步提升的方向。”该分析师说。
如何从困境中突围?
根据弗若斯特沙利文的资料,中国HIV药物市场由2017年的242.0百万美元增长至2021年的393.7百万美元,复合年增长率为12.9%。预期到2025年及2030年将分别达至11亿美元及27亿美元,2021年至2025年的复合年增长率为28.9%,2025年至2030年的复合年增长率为19.8%,远高于同期全球HIV药物市场6.0%及3.7%的预计复合年增长率。
中国市场上的大部分HIV药物为单剂抗逆转录病毒药物,而不是在发达市场更容 易获得的含有多种ART药物的复合药物。由于作为NRTI(广泛用作一线联合ART方案中的骨干药物的一种抗逆转录病毒药物),阿兹夫定会与不同机制的药物联合作用以形成各种ART方案,故不同类别的单剂抗逆转录病毒药物不被认为是阿兹夫定的替代品或竞争产品。
另据弗若斯特沙利文的资料,虽然2021年已有多种COVID-19疫苗获批,但仍可能需要新治疗方案以抗击COVID-19、减轻症状及加快患者康复进程。COVID-19目前可用的治疗方案包括中和抗体和抗病毒药物。截至2021年12月31日,全球已有约50家公司将治疗COVID-19的抗病毒候选药物推进至临床开发阶段。
平安证券研报指出,国内外各大企业也通过合作研发以及授权引入等方式对药物管线进行布局以快速抢占赛道。东吴证券也指出,需特别关注研发与商业化的确定性,即临床需求的紧迫性以及注册性临床数据,同时甄别能够带来营业收入的“收入管线”和用于提高估值的“花瓶管线”。
也是由于市场竞争焦灼、研发及商业化挑战不断,背后的风险也亟待关注。真实生物在申请书中称,公司已将风险及不确定因素分类为:与开发管线产品有关的风险;与产品制造及商业化有关的风险;与财务状况及额外资本需求有关的风险;与广泛的政府监管有关的风险;与知识产权有关的风险;与依赖第三方有关的风险;与行业及业务运营有关的风险;与在中国开展业务有关的风险等。
例如,临床药物开发过程漫长、成本高昂且结果充满不确定性,而前期研究及试验的结果未必能预示未来的试验结果。临床试验成本高昂,完成试验可能耗费多年时间,而其结果本身充满不确定性且不一定有利。临床试验的过程中随时可能出现失败。真实生物候选药物的临床前研究或早期临床试验结果未必能预示较后阶段的临床试验结果,且最初或中期试验结果未必能预示最终结果。尽管已通过临床前研究及初期临床试验,但处于临床试验较后阶段的候选药物可能无法展示出理想的安全性及疗效特性。
同时,在部分情况下,由于多种因素,同一候选药物的不同研究与试验之间,其安全性及/疗效结果亦可发生重大变化,该等因素包括试验方案所载试验程序出现变化、患者人群的人数及类别差异(包括性别 差异)、患者对给药方案的遵守程度及其他试验方案因素,以及临床试验参与者的退出率。有分析表明,进入后期临床开发的研究性药物可能在关键性临床试验期间或之后失败,主要是由于对安全性、疗效或两者的担忧。公司未来的临床试验结果未必理想。
此外,候选药物的临床试验未能展示令监管机构信纳的安全性及功效或未能另行产生正面的结果,也可能会产生额外成本或推迟完成或可能最终无法完成候选药物的开发及商业化。例如,公司未能达成获批后规定,获得国家药监局对阿兹夫定的附条件批准或会遭撤回。
根据公开信息,真实生物分别于2021年7月及2022年7月自国家药监局获得阿兹夫定治疗HIV感染及COVID-19的附条件批准。虽然真实生物获授权在中国销售用于治疗HIV感染的阿兹夫定,但在2026年续批阿兹夫定之前,公司须进行一项获批后III期临床试验,以监测阿兹夫定的疗效和安全性,并向国家药监局提交结果报告。同样地,虽然公司获准在中国销 售用于治疗COVID-19的阿兹夫定,但必须:
一是,开展阿兹夫定对SARS-CoV-2病毒突变变种的药效学研究;
二是,积极推进正在进行的阿兹夫定临床试验,并于完成后提交试验报告;
三是,持续收集批准后的有效性及安全性临床数据;
四是,自批准之日起三年内提交所需材料。概无法保证公司将能够及时完成获批后的临床研究或根本无法完成获批后的临床试验。
这也表示,如果真实生物未能定期提交安全报告及/或自批准日期起规定时限内提交临床试验报告,阿兹夫定的附条件批准可能遭撤回。鉴于上述,公司业务及未来发展或会严重受损,或会无法产生足够的收益和现金流继续营运,股份的市价可能会下跌。
如此,在较为可观的市场潜力下,面对市场风险重重,真实生物能否凭借在研以及已经商业化的药物实现盈利?值得关注。
(作者:季媛媛 编辑:徐旭)
21世纪经济报道及其客户端所刊载内容的知识产权均属广东二十一世纪环球经济报社所有。未经书面授权,任何人不得以任何方式使用。详情或获取授权信息请点击此处。
